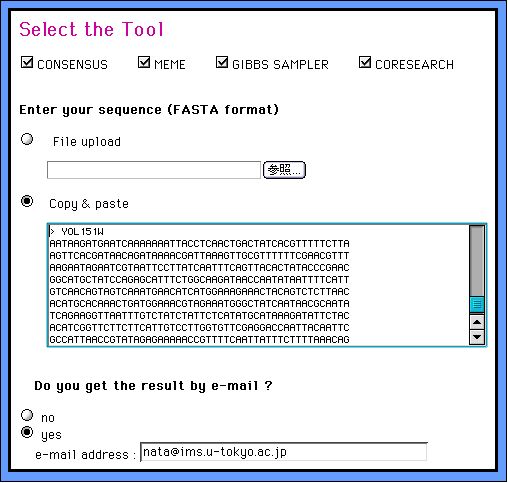
Select the Tool, Input & Submit the Dataset
We introduce here a novel Analyzer, called Melina,
whose main purpose is to elucidate effectively consensus
motif in a set promoters of co-regulated genes.
Four progressive motif extraction programs are included and offered for simultaneous usage in Melina, and their results can be observed and compared at a glance from the graphical output, called
"Comparison map", from the text format file "Motif Table Page" and in
a raw data format. Only run and compare!
All of the involved programs elucidate the consensus motif without any a priori knowledge
about its characteristics, although the motif length is presumed. We insist that it is possible to make these algorithms more
sensitive to the solution of various elucidation tasks in the range of biologically possible, such as, for example, elucidation of
single conserved motif or multiple corrupted motifs,
if apply the appropriate parameters combinations. As the output results are strongly dependent on the parameter set used, we carefully describe each parameter and its role in the algorithm. Also we give here some samples of different usage of parameters depending on the elucidation task.
In the case only one program is chosen from Melina, you can concomitantly apply 5 datasets and compare the results at a glance.
Here you will find a very friendly tutorial, which will teach you how to input a sequences set into the
Melina web page, choose the appropriate parameters, and examine the results. Good luck
to you!
1. Choose the program you want to use by switching on (off) the checkboxes, input your FASTA format set of DNA sequences and submit the dataset. If you use all programs, it looks like below:
After you submit the dataset you will go to the parameters setting page. In the case you choose only one program, you can set up to 5 parameter sets samples, i. e., to obtain 5 resulting outputs simultaneously for one dataset as it is shown below (CASE 1). Otherwise, only one parameters sets sample will be provided for each algorithm (CASE 2).
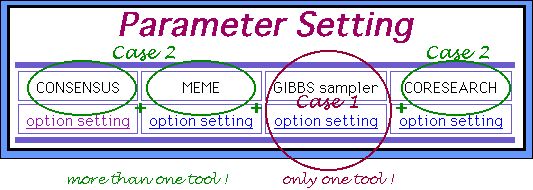
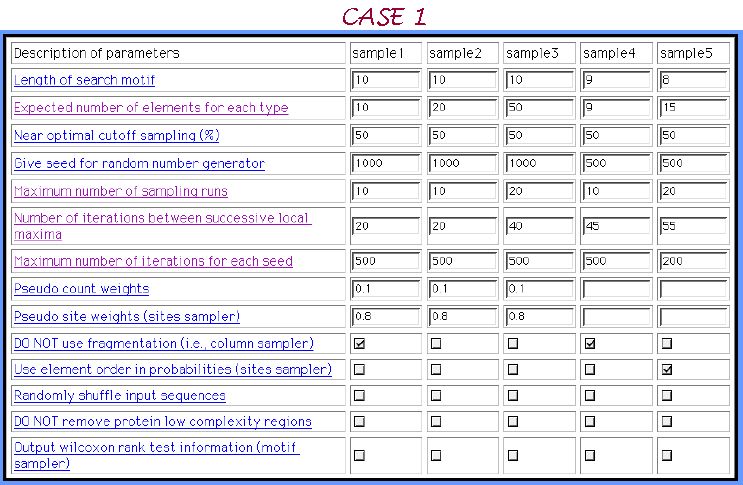
About CASE 1....
As an example here we choose only Gibbs sampler and apply to it the dataset of 30 randomly shuffled sequences, where a half of them possess per
3 inserted motifs with 3 mismatches in each motif. 5 samples of parameters sets are composed below:
Looking at the graphical view of the output, which called in our Melina as a Comparison map, and comparing these 5 samples we recognize that the parameters composition of sample 3 is the most appropriate one for the elucidation of multiple weak motifs.
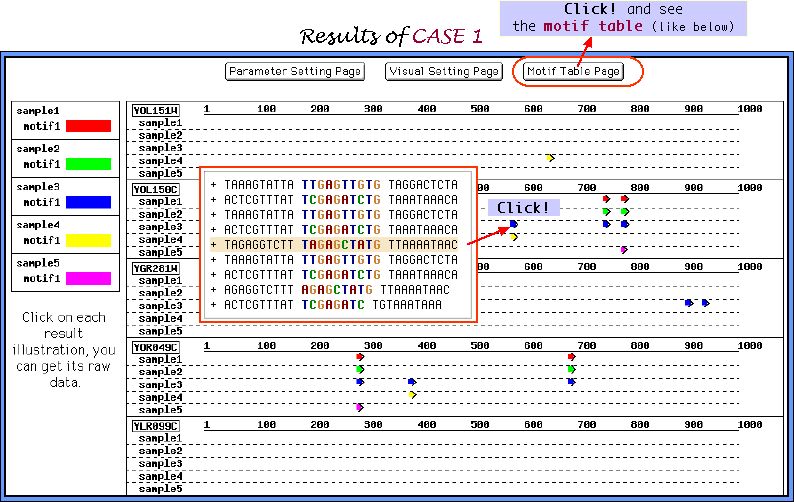
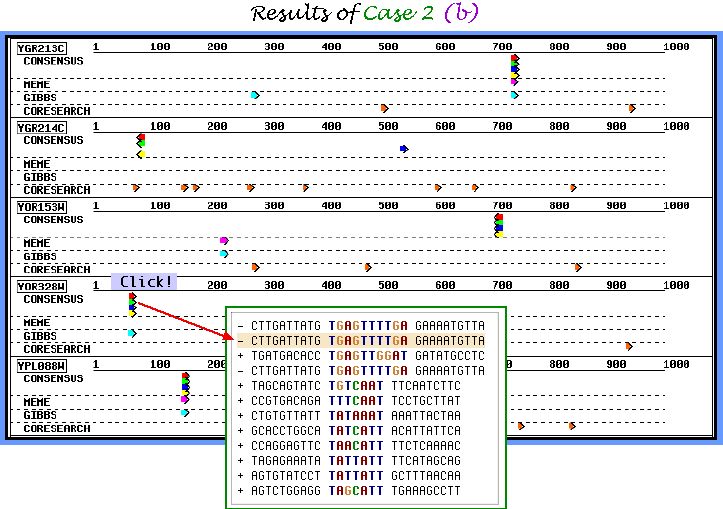
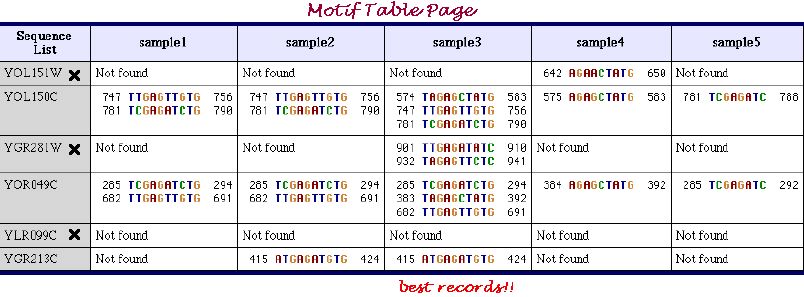
Motif Table Page shown below very conveniently represents the motifs extracted in all samples of parameters sets. Cross mark near the entry name indicates the sequence, which does not include the inserted motifs. As it is seen from the table, elucidation under the sample 3 of the parameters sets was the most successfull.
.
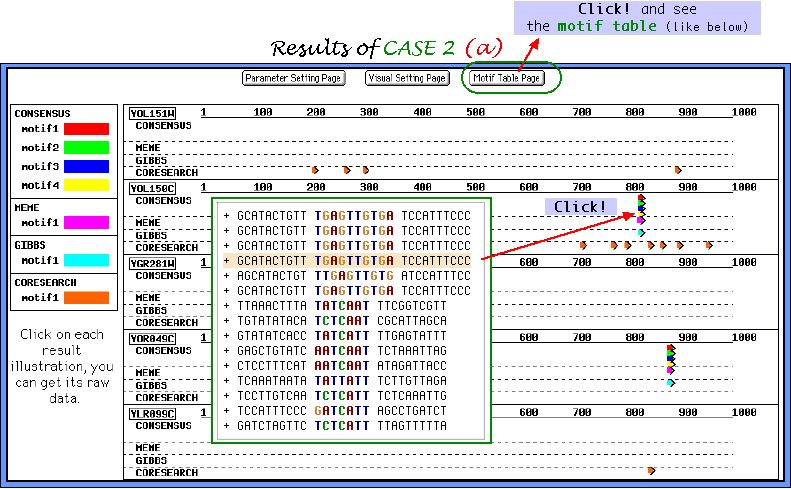
About CASE 2....
In the case you choose more than one tool, only one sample of parameters sets for each tool will be offered, like it is shown below. Here we demonstrate two examples of parameters sets for Consensus and two completely different outputs, emphasizing once again the importance of correct parameters usage depending on elucidation task. Other algorithms are used in a default mode. As a dataset here we use 30 nucleotide sequences, where half of them contains a single randomly inserted motif.
We are conferring two elucidation tasks here:
( a ) to extract single conserved motifs from the sequences where there are exist
( b ) to extract multiple weakly conserved motifs from the sequences they are exist

First, we report the graphical output of CASE 2 task (a). Almost all inserted motifs were successfully picked up by 3 out of 4 algorithms. Note, that no undesirable corrupted motifs were found by Consensus, and even Gibbs sampler and MEME do not show the ambiguous results.
Graphical output of CASE 2 task (b) parameters set is demonstrated below. This particular parameters combination allows to obtain a large number of similar weakly conserved motifs. We suppose it to be useful for finding motifs with mismatch level of 30-40%.